New paper published in Aggregate!
July 6, 2026
MetaSimulation of NonEquilibrium Processes
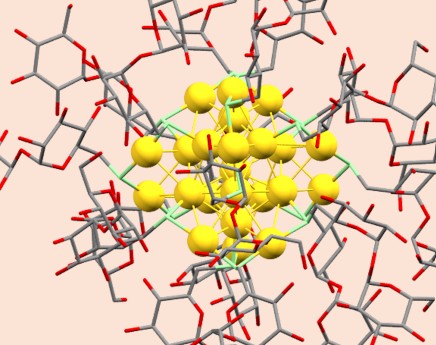
Atomically precise nanoclusters are key to materials, energy, and biomedical applications. Incorporating bioactive ligands has proven to be a significant challenge. In this work, we develop a straightforward method for the synthesis of an atomically precise gold nanocluster capped with a carbohydrate ligand, trehalose, Au25Tre18, in one step and in 55% yield. To confirm the structure and shed light on the nanocluster properties, we carry out a set of experiments, including high-resolution mass spectrometry, optical absorption spectroscopy, nuclear magnetic resonance spectroscopy (NMR), as well as theoretical calculations (DFT and TD-DFT). Furthermore, Au25Tre18 exhibited enhanced antimicrobial activity against Mycobacterium smegmatis.
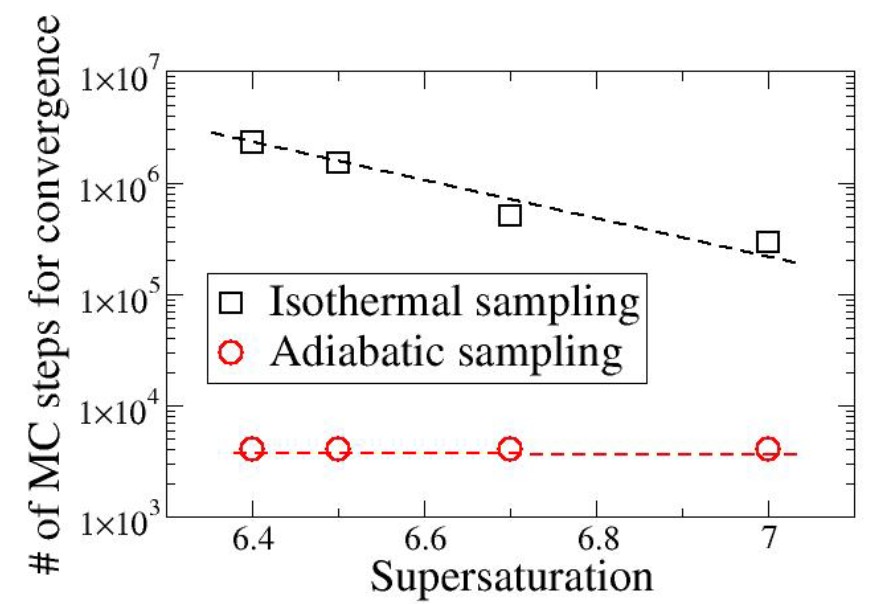
The efficient computational screening of large libraries of porous materials for gas storage applications relies on the ability of simulations to rapidly predict adsorption properties and selectivity in separation applications. While isothermal grand-canonical simulations are commonly used, their convergence rate can dramatically decrease when the thermodynamic conditions approach coexistence, giving rise to metastability and large free energy barriers. To address this challenge, we develop an adiabatic counterpart for the simulation of mixtures, derive the acceptance rules for a grand-isochoric adiabatic Monte Carlo method, and assess its accuracy and efficiency. The results show that adiabatic simulations allow for large temperature variations, thereby resulting in the rapid sampling of the configuration space and faster convergence.

We are excited to share some fantastic news! Arthea will be heading to Harvard Medical School to pursue a graduate degree in data science, while Kithma will be joining Genentech this summer as an intern. Kithma was also recently named a finalist in the 3MT competition. Congratulations to Arthea and Kithma on these well-deserved accomplishments!
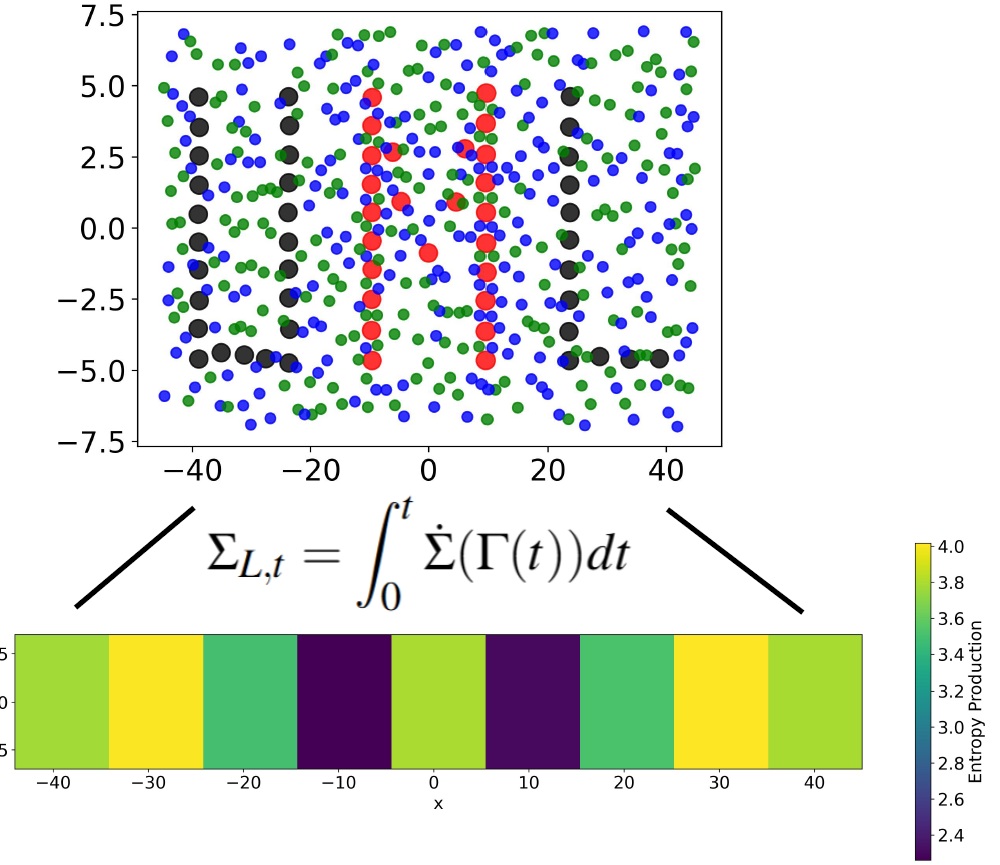
Dissipation in soft matter and living systems is highly heterogeneous, giving rise to complex local entropy production patterns. Recent work has identified the connection between local entropy production and locally extractable amount of work. This suggests that locally produced entropy can be recovered to power nanorobots and biological machines, prompting the need for efficient strategies to modulate and fine-tune local entropy production. In this work, we propose to program entropy production hotspots by leveraging local interaction patterns and test this approach in simulations of a fluid driven through a nanopore or past an obstacle.

The design of next-gen materials has undergone remarkable progress in recent years, as evidenced by the emergence of automated platforms combining artificial intelligence (AI)-driven synthesis planning and robotics for execution. In this Mini-Review, we analyze how synergistic approaches that combine driven quantum dynamics, AI/machine learning, and quantum computing accelerate the discovery and design process of quantum materials with enhanced properties and novel functionalities. We examine recent successes in next-gen materials science for quantum batteries, colloidal quantum dots solar cells, quantum phototransistors, rare-earth-free materials, and applications in quantum information processing.
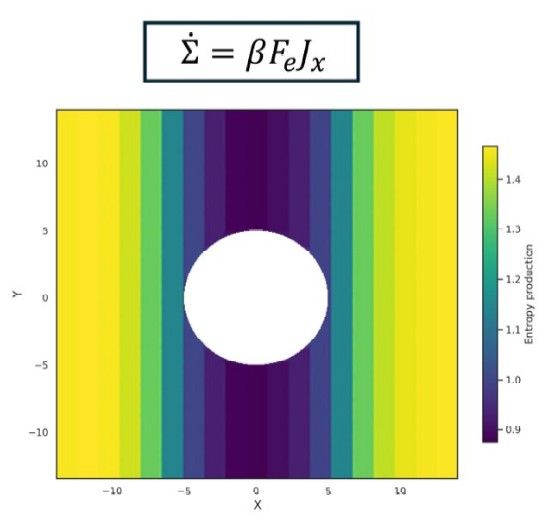
While global entropy production provides a measure of irreversibility, its partitioning into contributions from local regions is key to understanding the mechanisms underlying time-reversal symmetry breaking in complex systems and active matter. Here, by analyzing local heat flows and fluxes, we propose a framework that enables the mapping of local dissipation and entropy production in a nonequilibrium system. We test this approach in simulations of fluids driven through complex environments and active systems. We connect the results across the local and global scales by showing that local dissipation and entropy production satisfy a local version of the usual (global) fluctuation theorem, which accounts for the correlations between the local region and its surroundings.
Predicting the outcome of a crystallization process remains a long-standing challenge in solid state chemistry. This stems from the subtle interplay between thermodynamics and kinetics that results in a complex crystal energy landscape, spanned by many polymorphs and other metastable intermediates. Molecular simulations are uniquely positioned to unravel this interplay, as they constitute a framework that can compute free energies (thermodynamics), barriers (kinetics), and visualize the crystallization mechanisms at high resolution. We show here how recent progress in computational methods, and their augmentation with Machine Learning, has advanced our ability to predict crystal structure and simulate crystal nucleation.
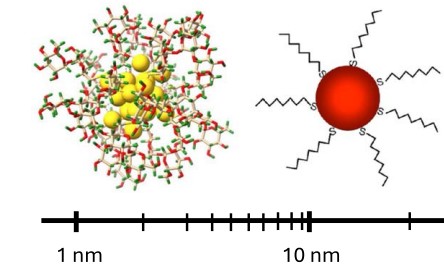
Gold nanoparticles, and their smaller (< 2 nm) counterpart, known as gold nanoclusters, have emerged in recent years as highly efficient catalysts, and are highly promising for applications in nanomedicine, sensing, and bioimaging. Here we discuss how Machine-Learned Potentials have led to the elucidation of the structure, stability, thermodynamics, and reactivity of nanomaterials, thereby paving the way for the accelerated computationally-guided design of Au nanomaterials.

Computations, AI, and ML were in the news with the award of the 2024 Nobel Prize in Physics to John J. Hopfield and Geoffrey E. Hinton, and with the award of teh 2024 Nobel Prize in Chemistry to David Baker, Demis Hassabis, and John M. Jumper. You can read more about the awards and Dr. Delhommelle's reaction in Scientific American and in the Wall Street Journal.
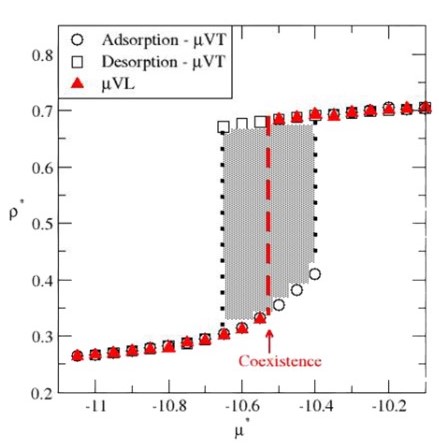
Under isothermal conditions, phase transitions occur through a nucleation event when conditions are sufficiently close to coexistence. This leads to a very slow kinetics for the transition, ultimately resulting in hysteresis, where the system can remain in a metastable state for a long time. This has broad implications, for instance, when using simulations to predict phase diagrams or screen porous materials for gas storage applications. Here, we leverage simulations in an adiabatic statistical ensemble, known as adiabatic grand-isochoric ensemble ensemble, to reach equilibrium states with a greater efficiency than its isothermal counterpart, i.e., simulations in the grand-canonical ensemble.

The book builds on the analogy between social groups and assemblies of molecules to introduce the concepts of statistical mechanics, machine learning and data science. Applying a data analytics approach to molecular systems, we show how individual (molecular) features and interactions between molecules, or "communication" processes, allow for the prediction of properties and collective behavior of molecular systems - just as polling and social networking shed light on the behavior of social groups. Applications to cutting-edge research for biological, environmental, and energy systems are also presented.
Developing new pharmaceutical compounds is a lengthy, costly, and intensive process. In recent years, the development of Artificial Intelligence (AI), Machine Learning (ML), and Deep Learning (DL) models has drawn considerable interest in drug discovery. In this review, we discuss recent advances in the field and show how these methods can be leveraged to assist each stage of the drug discovery process.
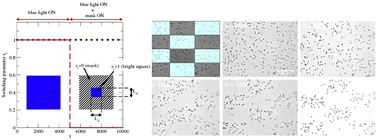
The ability of active matter to assemble into reconfigurable nonequilibrium structures has drawn considerable interest in recent years. We investigate how active fluids respond to spatial light patterns through simulations and experiments on light-activated self-propelled colloidal particles. We examine the processes of inverse templated assembly, which involves creating a region without active particles through a bright pattern, and templated assembly, which promotes the formation of dense particle regions through a dark pattern. We identify scaling relations for the characteristic times for both processes that quantify the interplay between the dimension of the applied pattern and the intrinsic properties of the active fluid.

The past years have been marked by fast-paced advances in the data-driven and Machine Learning (ML)-aided design and characterization of energy materials. Equally remarkable is the broad range of theoretical methods, physical systems, and applications that ML-aided research has enabled to explore. This symposium will bring together chemists, physicists, and engineers from these different areas, with complementary computational and experimental expertise, to share the cutting-edge advances in the design of energy materials enabled by ML in their respective fields. Join us for this exciting symposium co-organized with Mingda Li (MIT) and Fang Liu (Emory)

Understanding and optimizing solid-solid interfaces in graphene-based nanocomposite catalysts is crucial for electrocatalytic applications such as CO2 reduction. Here we develop a new strategy for constructing innovative electrocatalysts from metal nanoclusters covalently attached onto three-dimensional pristine graphene electrodes through rationally designed molecular linkers. Using a combination of synthesis, computational modeling, electrochemistry, and spectroscopy, we show how the control over solid-solid interfaces impacts performance in electrochemical devices. Stay tuned for more news and work with our collaborators Kwok-Fan Chow and Mingdi Yan at UML and Gonghu Li at UNH.
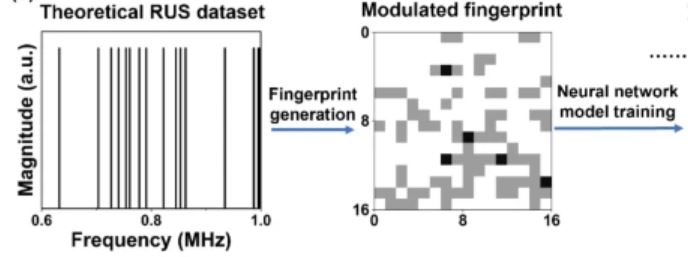
Read the paper here. A modulated fingerprint assisted machine learning method for retrieving elastic moduli from resonant ultrasound spectroscopy. We used deep-learning-based models to automatically obtain elastic moduli from resonant ultrasound spectroscopy (RUS) spectra, which conventionally require user intervention of published analysis codes. By strategically converting theoretical RUS spectra into their modulated fingerprints and using them as a dataset to train neural network models, we obtained models that successfully predicted elastic moduli. In summary, our modulated fingerprint method is an efficient tool to transform raw spectroscopy data and train neural network models with high accuracy and resistance to spectra distortion.

Living systems have the unique ability to form hierarchical assemblies, in which individual constituents can perform tasks cooperatively and emergently. Harnessing such properties is a long-standing challenge for the rational design of dynamic materials, that can respond to their environment, communicate with one another, and undergo a rapid, reversible, assembly through the transduction of energy. Here we develop a combined experimental, computational, theoretical and Machine Learning framework to program the assembly of smart active materials. Stay tuned for more news and work with our collaborators Paul Chaikin, Stefano Sacanna and Mark Tuckerman at NYU.

Molecular crystals play an essential role in the pharmaceutical, agrochemical, electronics, and defense industries. In many instances, a given chemical compound may have more than one crystal structure, a phenomenon known as polymorphism. A crystal may also contain impurities, the most important among these being water. Such structures are referred to as crystal hydrates. The ability of these materials to function in a desired manner may depend on which structure, pure or impure, they form. Utilizing advances in high-performance computing and artificial intelligence, we create new computational approaches and software components for rapidly predicting polymorphic structures in molecular crystals and understanding the transitions between structures. More info about our latest work with our collaborator Mark Tuckerman at NYU very soon.