MetaSimulation of Nonequilibrium Processes
The MSNEP Group
Journal Publications
2026
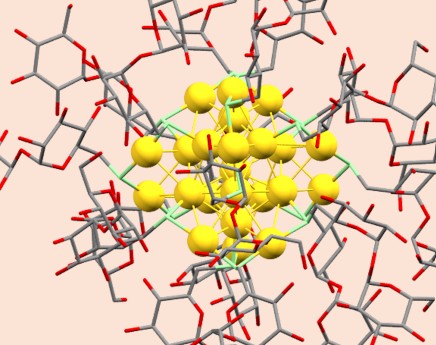
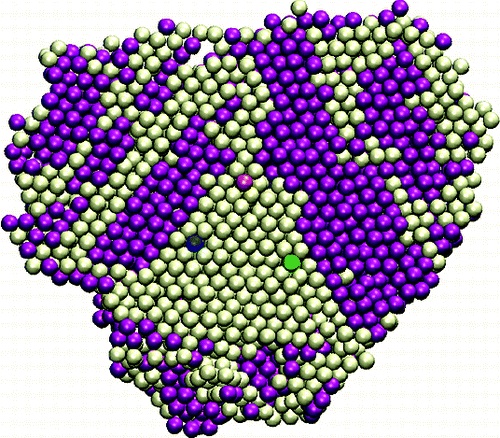
Atomically Precise Trehalose-Capped Au25 Nanoclusters: Synthesis and Tunable Antimicrobial Activity
M. Bodiuzzaman, H. A. Perera, D. Truong, J. Tu, W. Ndugire, W. E. Gavin, K. Sajini, J. Delhommelle, and M. Yan
Aggregate, 7, e70396 (2026)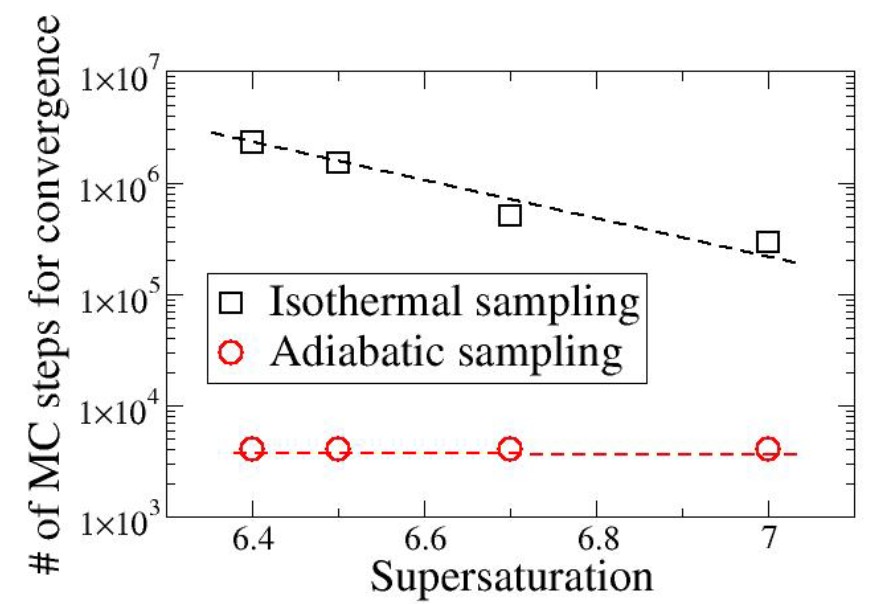
Rapid Exploration of Mixture Adsorption via Adiabatic Sampling
C. Desgranges and J. Delhommelle
J. Phys. Chem. C 130, 9180 (2026)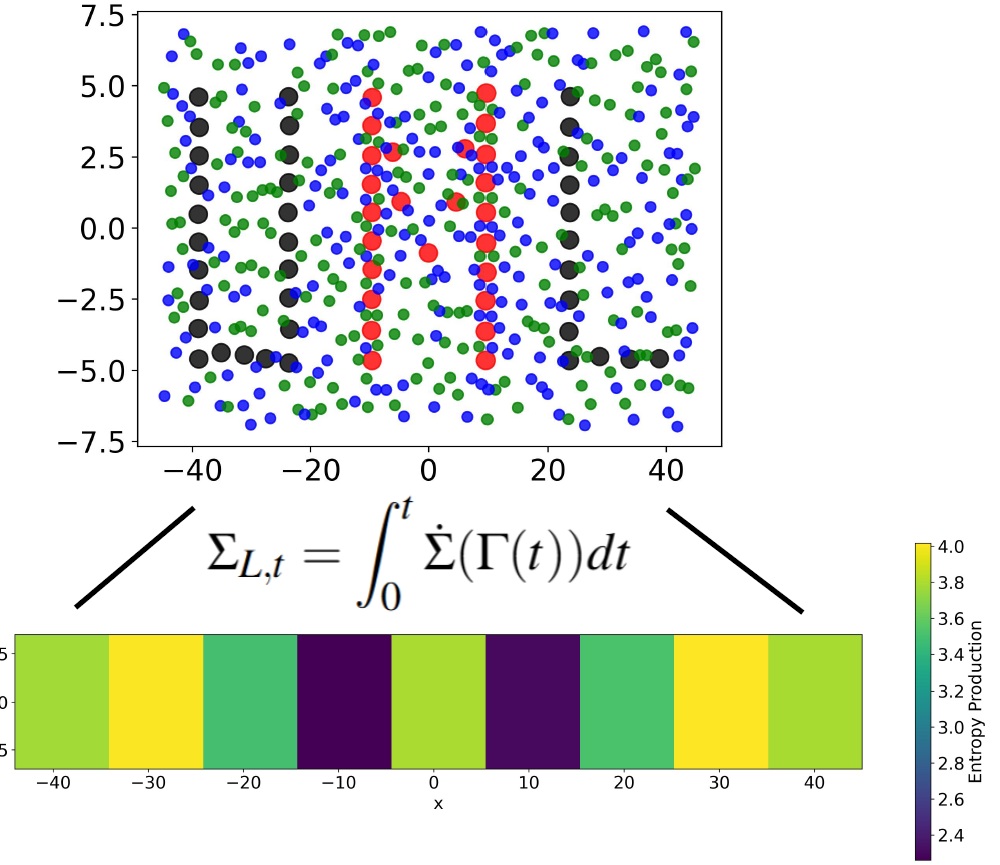
Programming entropy production hotspots via interaction patterning
C. Desgranges and J. Delhommelle
Soft Matter 22, 1655 (2026)2025

Mini Review: Synergizing Driven Quantum Dynamics, AI, and Quantum Computing for Next-Gen Materials Science
O. S. Akanbi, J. P. Shannon, J. Delhommelle, and C. Desgranges
J. Phys. Chem. Lett. 16, 11821 (2025)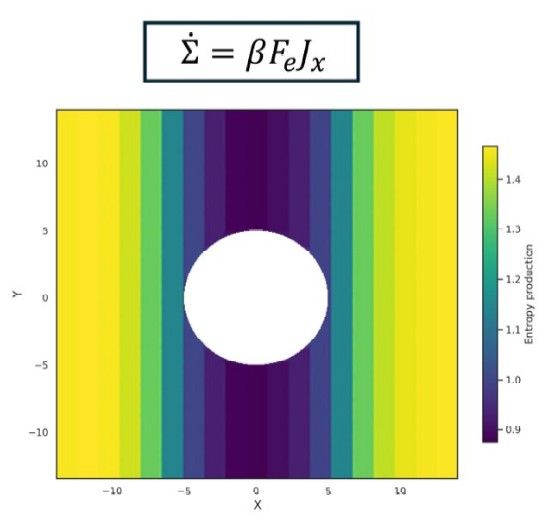
Mapping Local Dissipation and Entropy Production in Complex and Active Fluids
C. Desgranges and J. Delhommelle
J. Phys. Chem. Lett. 16, 11405 (2025)
Deciphering the complexities of crystalline state(s) with molecular simulations
C. Desgranges and J. Delhommelle
Commun. Chem. 8, 281 (2025)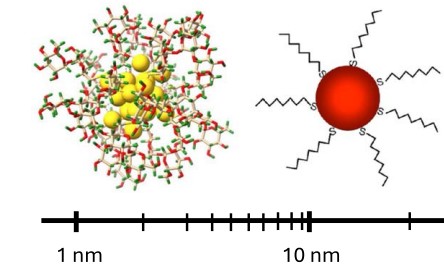
Advancing the design of gold nanomaterials with machine-learned potentials
K. Sajini, C. Desgranges, and J. Delhommelle
Nano. X 6, 022001 (2025)2024
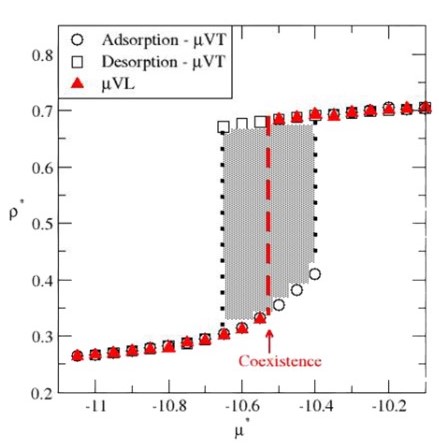
Accelerated convergence via adiabatic sampling for adsorption and desorption processes
C. Desgranges and J. Delhommelle
J. Chem. Phys. 161, 104104 (2024)Selected as an Editor's pick
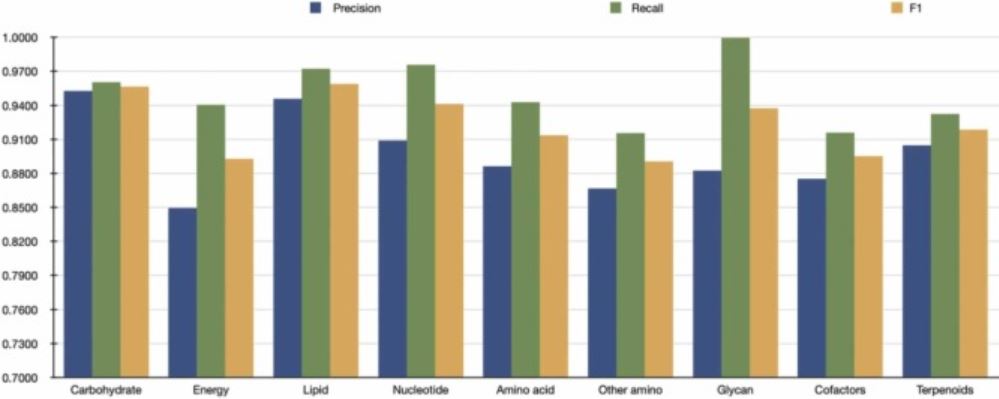
AI's role in pharmaceuticals: Assisting drug design from protein interactions to drug development
S. Bechelli and J. Delhommelle
Artificial Intelligence Chemistry 2, 100038 (2024)2023
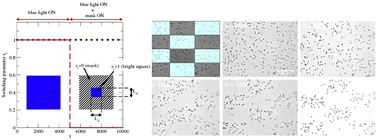
Microswimmers under the spotlight: interplay between agents with different levels of activity
C. Desgranges, M. Ferrari, P. M. Chaikin, S. Sacanna, M. E. Tuckerman, and J. Delhommelle
Soft Matter 19, 7334 (2023)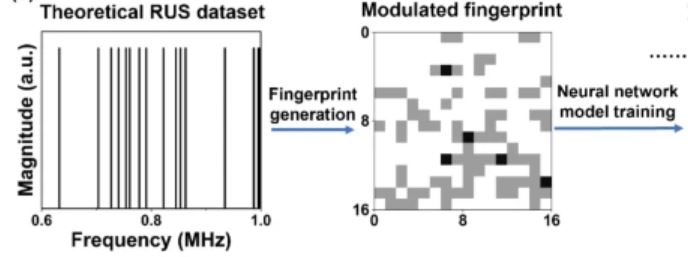
A modulated fingerprint assisted machine learning method for retrieving elastic moduli from resonant ultrasound spectroscopy
J. Liu, X. Zhao, K. Zhao, V. G. Goncharov, J. Delhommelle, J. Lin, and X. Guo
Sci. Rep. 13, 5919 (2023)2022
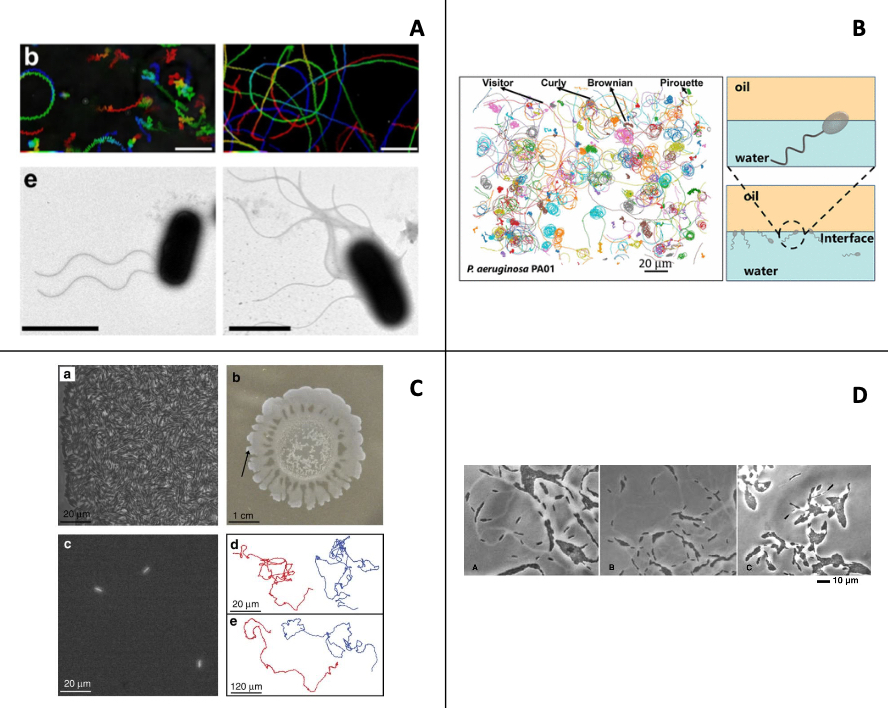
Designing, synthesizing, and modeling active fluids
I. Essafri, B. Ghosh, C. Desgranges, J. Delhommelle
Phys. Fluids 34, 071301, 2022
Entropy determination for mixtures in the adiabatic grand-isobaric ensemble
C. Desgranges, J. Delhommelle
J. Chem. Phys. 156, 084113, 2022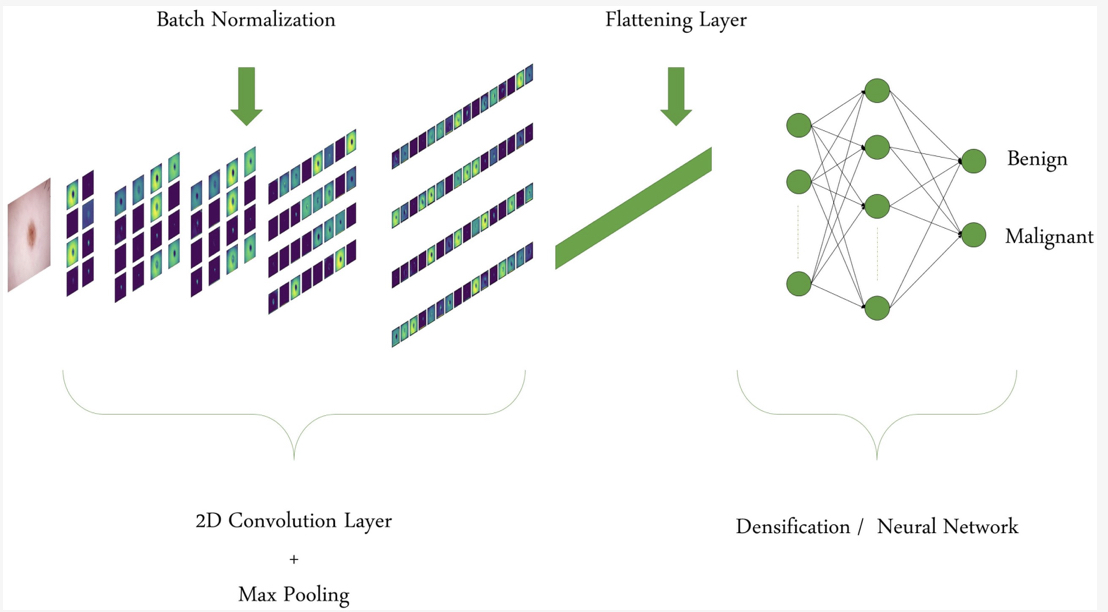
Machine Learning and Deep Learning Algorithms for Skin Cancer Classification from Dermoscopic Images
S. Bechelli, J. Delhommelle
Bioengineering 9, 97, 2022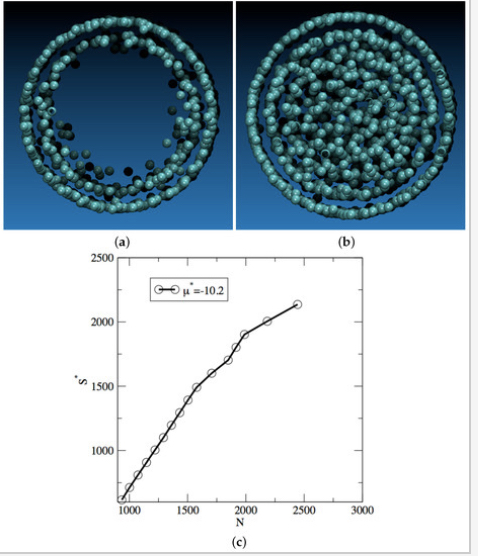
Machine-learned free energy surfaces for capillary condensation and evaporation in mesopores
C. Desgranges, J. Delhommelle
Entropy 24, 97, 20222021
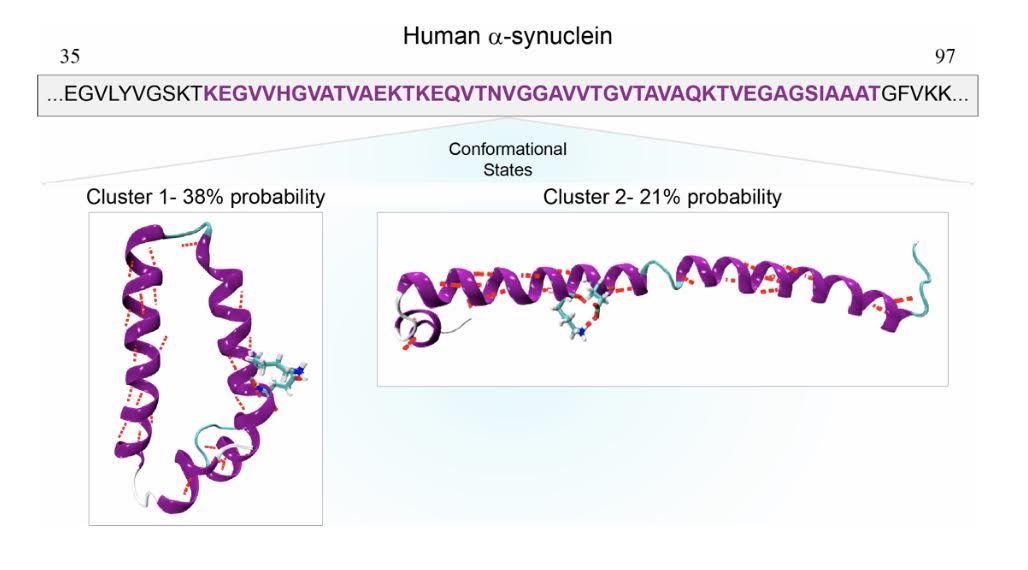
Identification of the folding landscape of a-synuclein (35-97) using replica-exchange molecular dynamics
K. Jain, O. Ghribi, J. Delhommelle
J. Chem. Inf. Model., 61, p. 432-443, 2021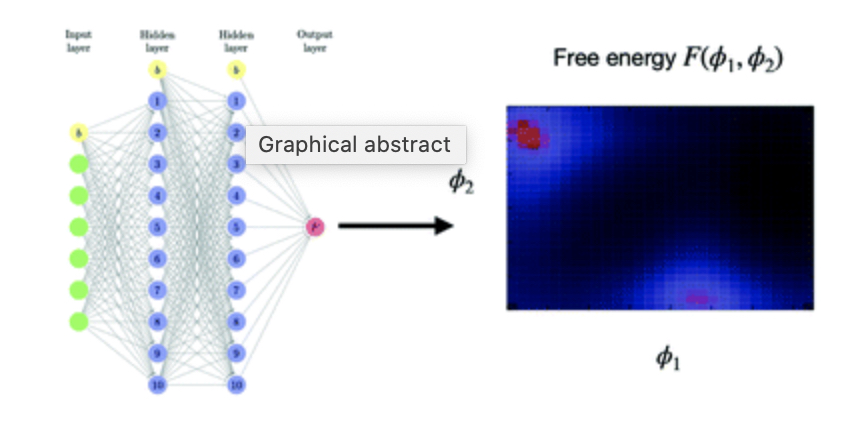
Towards a Machine Learned Thermodynamics: Exploration of Free Energy Landscapes in Molecular Fluids, Biological Systems and for Gas Storage and Separation in Metal-Organic Frameworks
C. Desgranges, J. Delhommelle
Mol. Syst. Eng. Des. Eng., 6, p. 52-65, 2021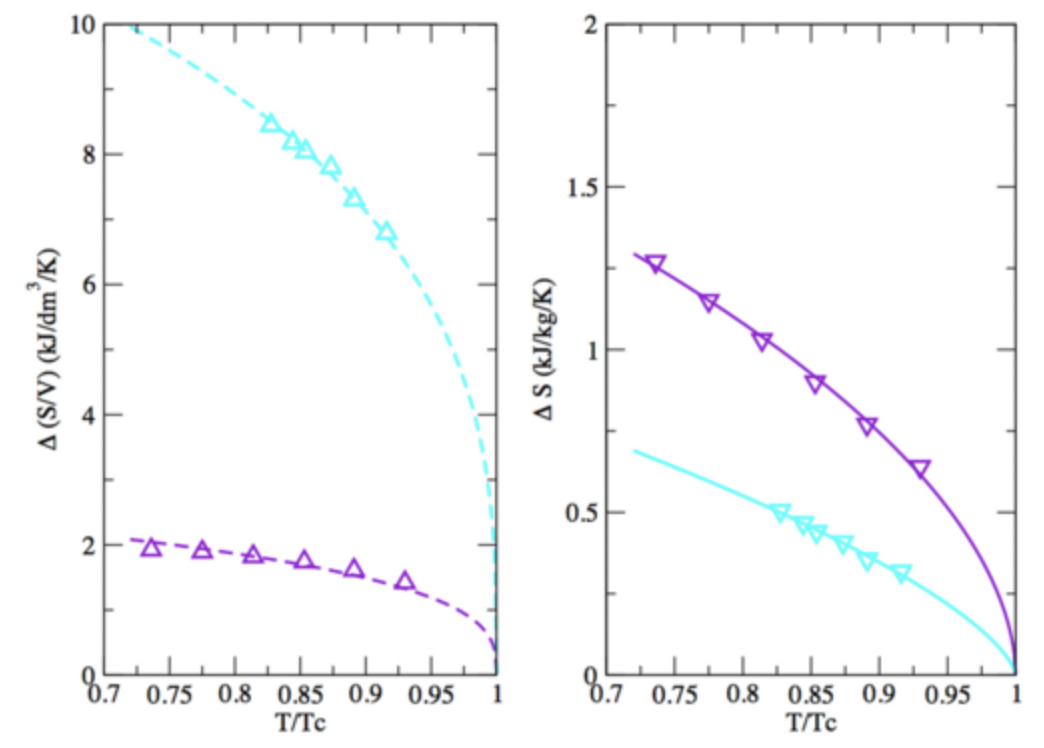
Entropy scaling close to criticality: From simple to metallic systems
C. Desgranges, J. Delhommelle
Phys. Rev. E, 103, 052102, 20212020
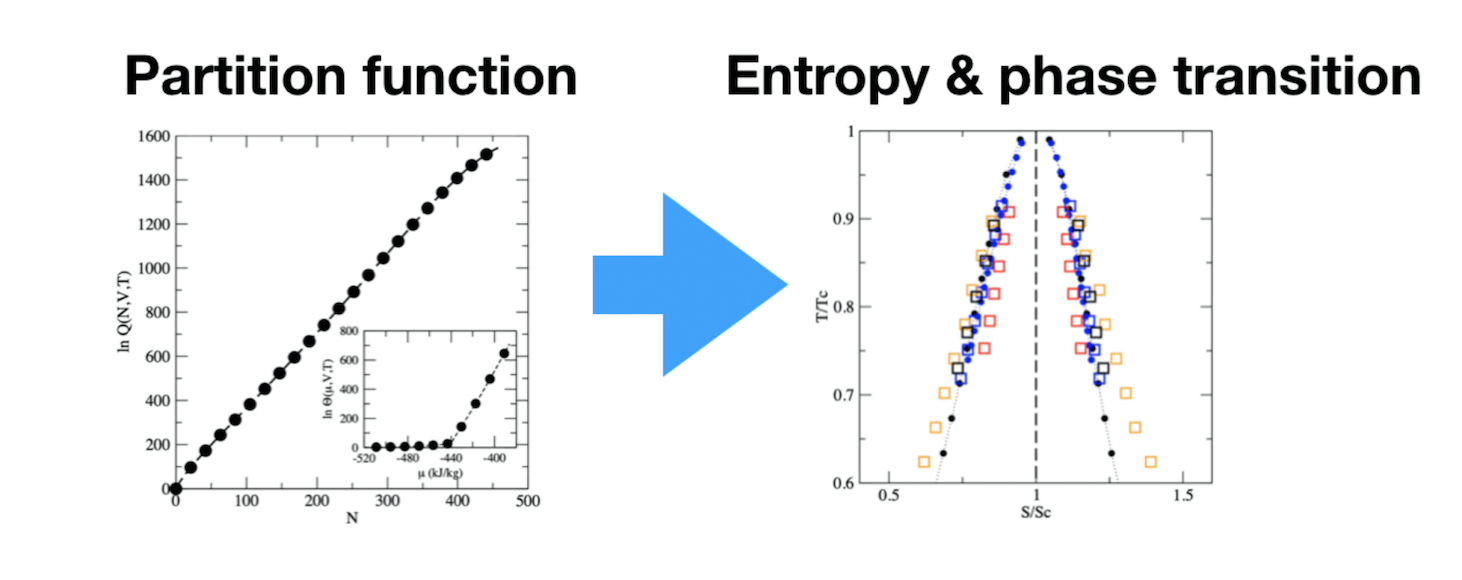
Entropy in Molecular Fluids: Interplay between Interaction Complexity and Criticality
C. Desgranges, J. Delhommelle
J. Phys. Chem. B, 124, p. 11463-11471, 2020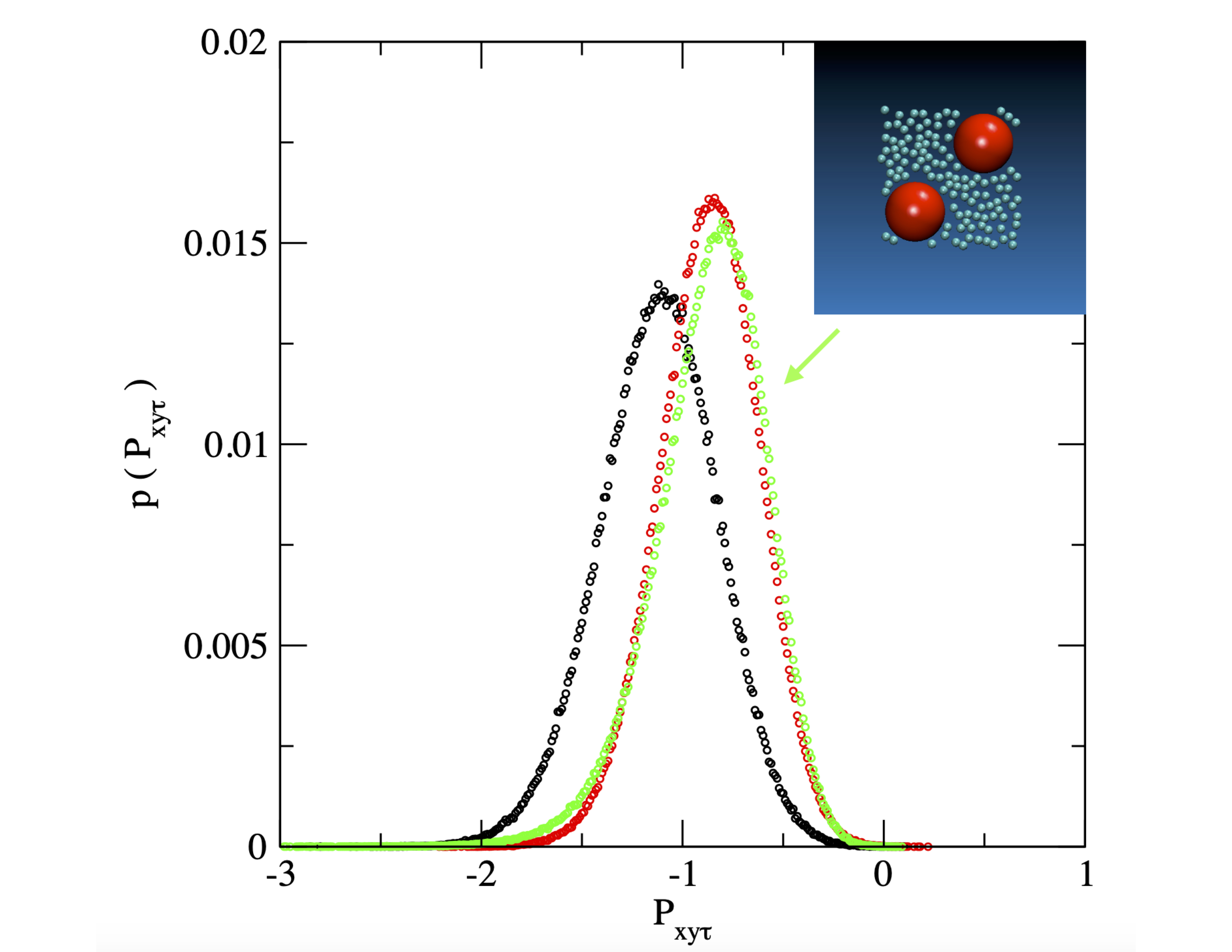
Entropy production in model colloidal suspensions under shear via the Fluctuation Theorem
C. Desgranges, J. Delhommelle
J. Chem. Phys., 153, p. 224113, 2020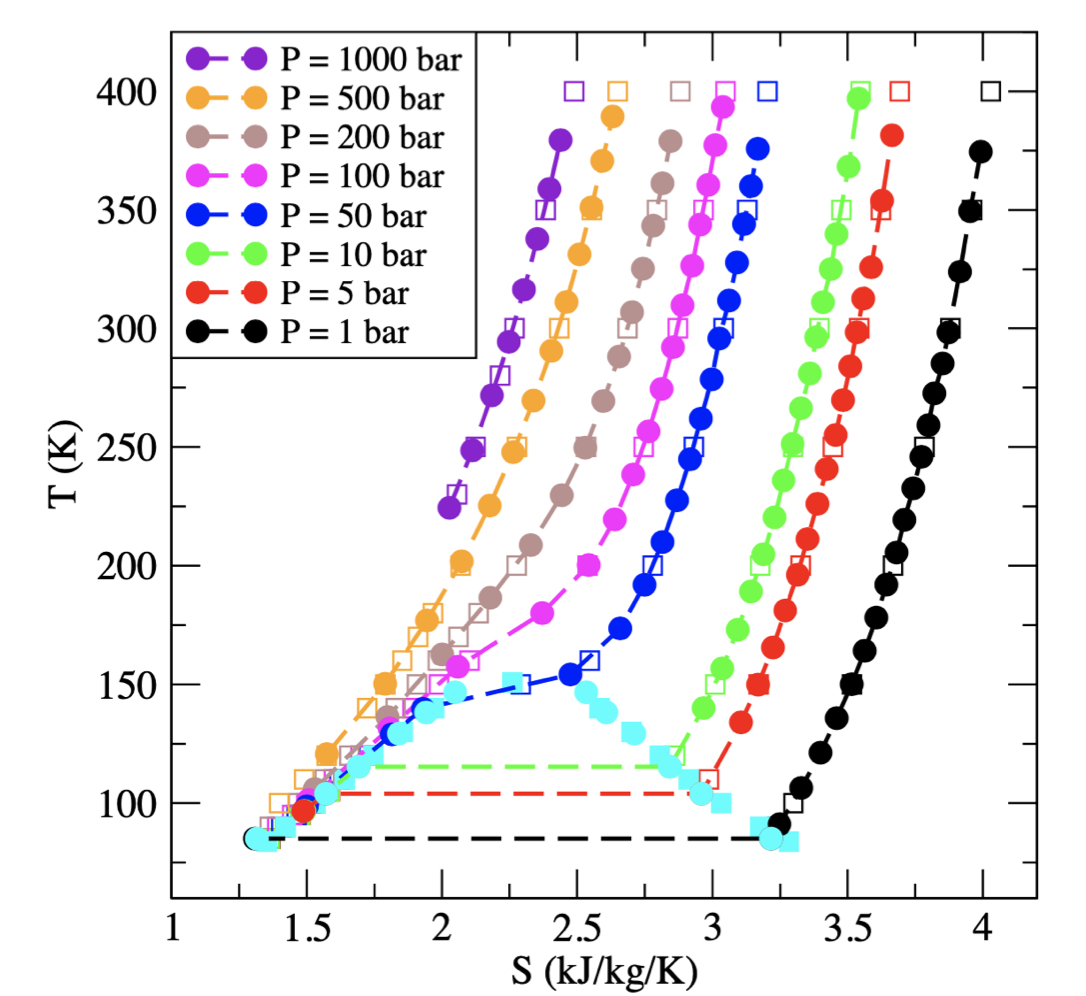
The central role of entropy in adiabatic ensembles and its application to phase transitions in the grand-isobaric adiabatic ensemble
C. Desgranges, J. Delhommelle
J. Chem. Phys., 153, p. 094114, 2020
Unraveling liquid polymorphism in silicon driven out-of-equilibrium
C. Desgranges, J. Delhommelle
J. Chem. Phys., 153, p. 054502, 2020
Ensemble Learning of Partition Functions for the Prediction of Thermodynamic Properties of Adsorption in Metal−Organic and Covalent Organic Frameworks
C. Desgranges, J. Delhommelle
J. Phys. Chem. C, 124, p. 1907-1917, 20202019

Can Ordered Precursors Promote the Nucleation of Solid Solutions?
C. Desgranges, J. Delhommelle
Phys. Rev. Lett., 123, p. 195701, 2019
Nucleation of Capillary Bridges and Bubbles in Nanoconfined CO2
C. Desgranges, J. Delhommelle
Langmuir, 35, p. 15401-15409, 2019
Stabilization of nanobubbles under hydrophobic confinement
C. Desgranges, J. Delhommelle
J. Phys. Chem. C, 123, p. 11707-117138, 2019
Prediction of the boiling and critical points of polycyclic aromatic hydrocarbons via Wang-Landau simulations and machine learning
S. D. Groven, C. Desgranges, J. Delhommelle
Fluid Phase Equil., 484, p. 225-231, 2019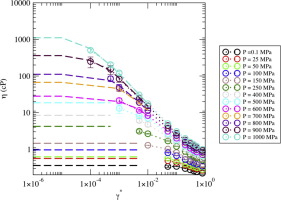
Viscosity of a highly compressed methylated alkane via equilibrium and nonequilibrium molecular dynamics simulations
I. Valencia-Jaime, C. Desgranges, J. Delhommelle
Chem. Phys. Lett., 719, p. 103-109, 2019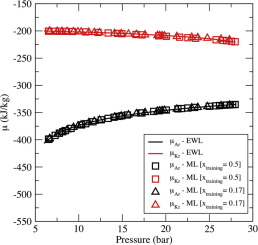
Determination of mixture properties via a combined Expanded Wang-Landau simulations-Machine Learning approach
C. Desgranges, J. Delhommelle
Chem. Phys. Lett., 715, p. 1-6, 20192018
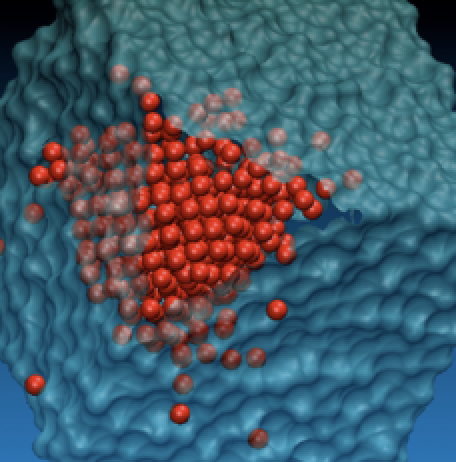
Crystal nucleation along an entropic pathway: Teaching liquids how to transition
C. Desgranges, J. Delhommelle
Phys. Rev. E, 98, p. 063307, 2018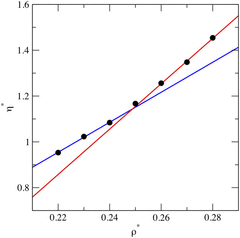
Communication: Existence and control of liquid polymorphism in methanol under shear
C. Desgranges, J. Delhommelle
J. Chem. Phys., 149, p. 111101, 2018
Prediction of the phase equilibria for island-type asphaltenes via HMC-WL simulations
C. Desgranges, J. Delhommelle
J. Chem. Phys., 149, p. 072307, 2018
A new approach for the prediction of partition functions using machine learning techniques
C. Desgranges, J. Delhommelle
J. Chem. Phys., 149, p. 044118, 2018
Modeling antigen-antibody nanoparticle bioconjugates and their polymorphs
C. Desgranges, J. Delhommelle
J. Chem. Phys., 148, p. 124507, 2018
Unusual Crystallization Behavior Close to the Glass Transition
C. Desgranges, J. Delhommelle
Phys. Rev. Lett., 120, p. 115701, 2018
Calculating free energy profiles using entropy as a reaction coordinate: Application to water nucleation
C. Desgranges, J. Delhommelle
Chem. Phys. Lett., 695, p. 194-199, 2018
Non-monotonic variations of the nucleation free energy in a glass-forming ultra-soft particles fluid
C. Desgranges, J. Delhommelle
Soft Matter, 14, p. 5977-5985, 20182017

Competition between crystalline and icosahedral order during crystal growth in bimetallic systems
B. Gonzalez, S. Bechelli, I. Essafri, V. Piquet, C. Desgranges, J. Delhommelle
J. Cryst. Growth, 478, p. 22-27, 2017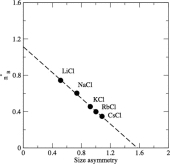
Similarity law and critical properties in ionic systems
C. Desgranges, J. Delhommelle
Chem. Phys. Lett., 687, p. 9-13, 2017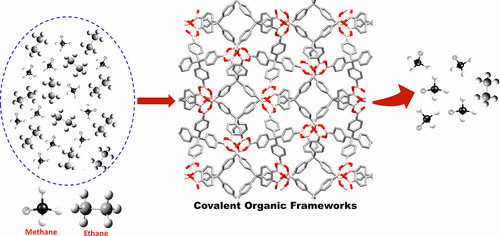
Selectivity and Desorption Free Energies for Methane–Ethane Mixtures in Covalent Organic Frameworks
K. Gopalsamy, C. Desgranges, J. Delhommelle
J. Phys. Chem. C, 121, p. 24692-24700, 2017
Benchmark Free Energies and Entropies for Saturated and Compressed Water
C. Desgranges, J. Delhommelle
J. Chem. & Engineering Data, 62, p. 4032-4040, 2017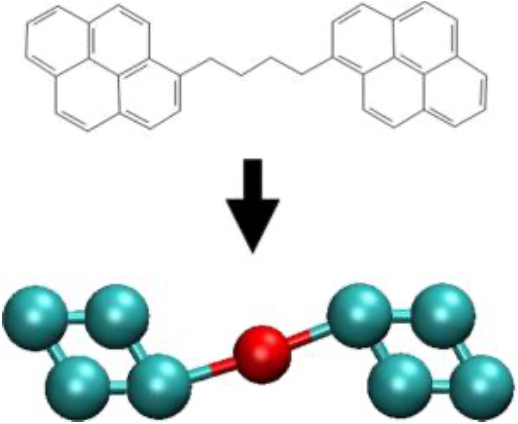
Coarse-Grained Model and Boiling Point Prediction for Asphaltene Model Compounds via HMC-WL Simulations
C. Desgranges, J. Delhommelle
Energy & Fuels, 31, p. 10699-10705, 2017
Free Energy of Nucleation and Interplay between Size and Composition in CuNi Systems
S. Bechelli, B. Gonzalez, V. Piquet, I. Essafri, C. Desgranges, J. Delhommelle
J. Phys. Chem. B, 121, p. 8558-8563, 2017
Free energy calculations along entropic pathways. III. Nucleation of capillary bridges and bubbles
C. Desgranges, J. Delhommelle
J. Chem. Phys., 146, p. 184104, 2017
Ginzburg-Landau free energy for molecular fluids: Determination and coarse-graining
C. Desgranges, J. Delhommelle
Chem. Phys. Lett., 669, p. 218-223, 2017
Classical and quantum many-body effects on the critical properties and thermodynamic regularities of silicon
C. Desgranges, P.W. Anderson, J. Delhommelle
J. Condens. Matter Phys., 29, p.045401, 20172016
Free energy calculations along entropic pathways. II. Droplet nucleation in binary mixtures
C. Desgranges, J. Delhommelle
J. Chem. Phys.,145, p. 234505, 2016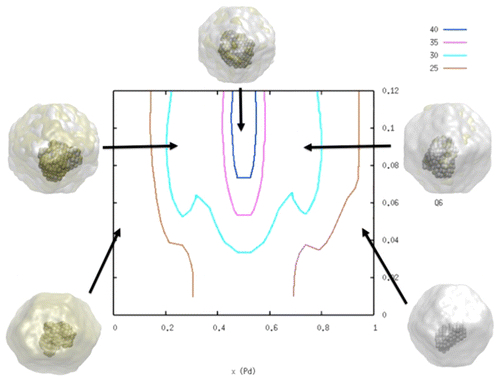
Effect of the Composition on the Free Energy of Crystal Nucleation for CuPd Nanoalloys
C. Desgranges, J. Delhommelle
J. Phys. Chem. C, 120 p. 27657-27664, 2016
Free energy calculations along entropic pathways. I. Homogeneous vapor-liquid nucleation for atomic and molecular systems
C. Desgranges, J. Delhommelle
J. Chem. Phys., 145 p. 204112, 2016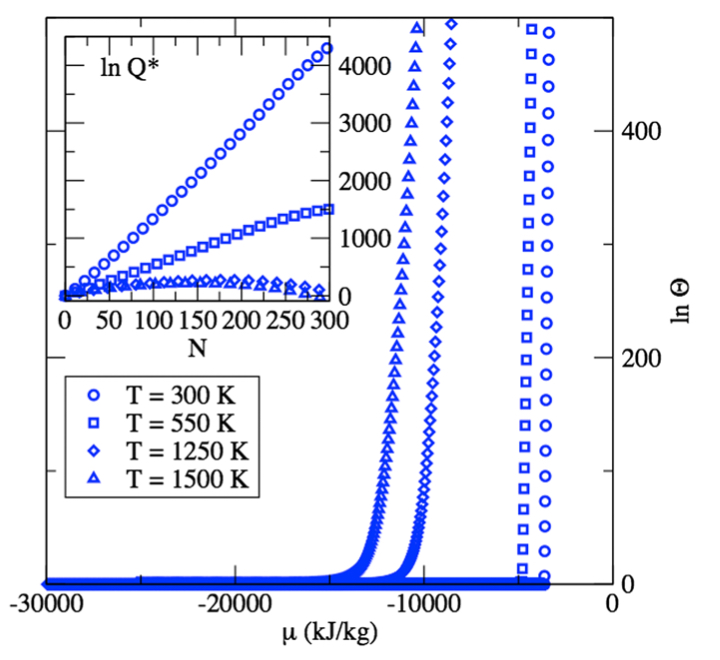
Evaluation of the grand-canonical partition function using expanded Wang-Landau simulations. V. Impact of an electric field on the thermodynamic properties and ideality contours of water
C. Desgranges, J. Delhommelle
J. Chem. Phys., 145 p. 184504, 2016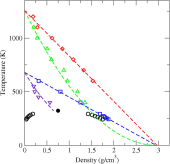
Ideality contours and thermodynamic regularities in supercritical molecular fluids
C. Desgranges, A. Margo, J. Delhommelle
Chem. Phys. Lett., 658 p. 37-42, 2016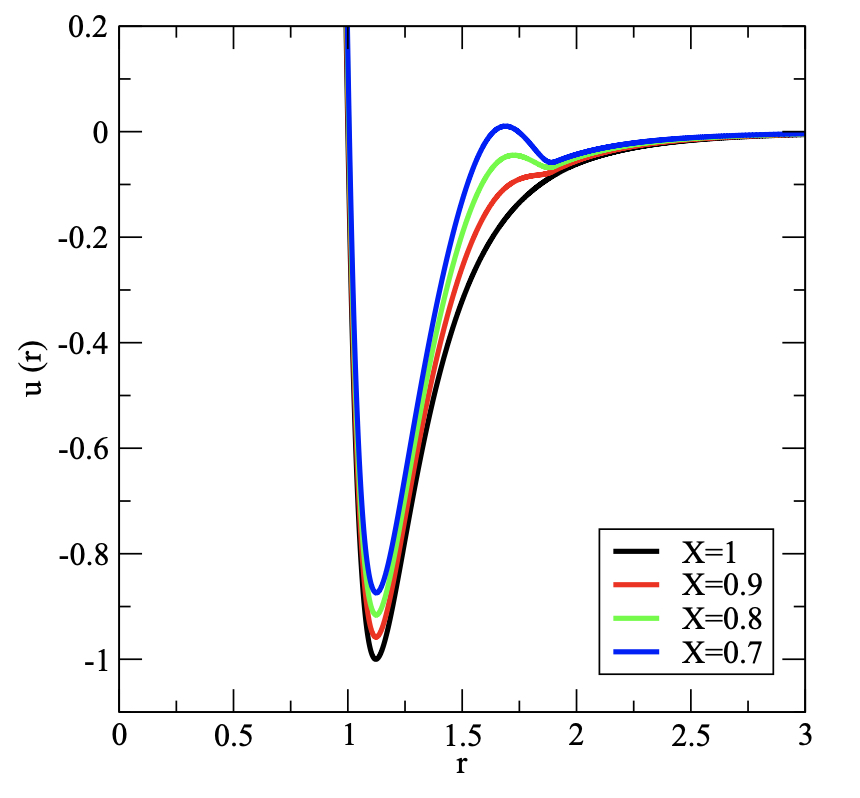
Impact of Friedel oscillations on vapor-liquid equilibria and supercritical properties in two and three dimensions
C. Desgranges, L. Huber, J. Delhommelle
Phys. Rev. E, 94 p. 012612, 2016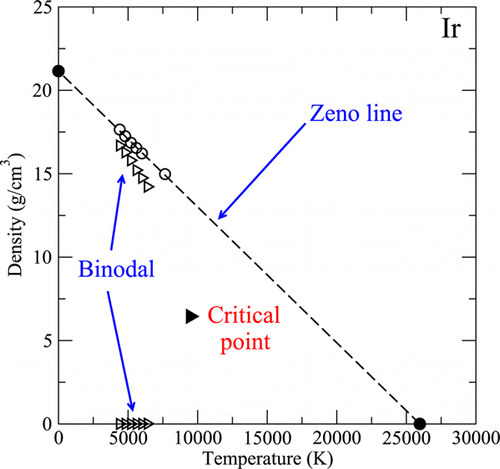
Scaling Laws and Critical Properties for fcc and hcp Metals
C. Desgranges, L. Widhalm, J. Delhommelle
J. Phys. Chem. B, 120 p. 5255-5261, 2016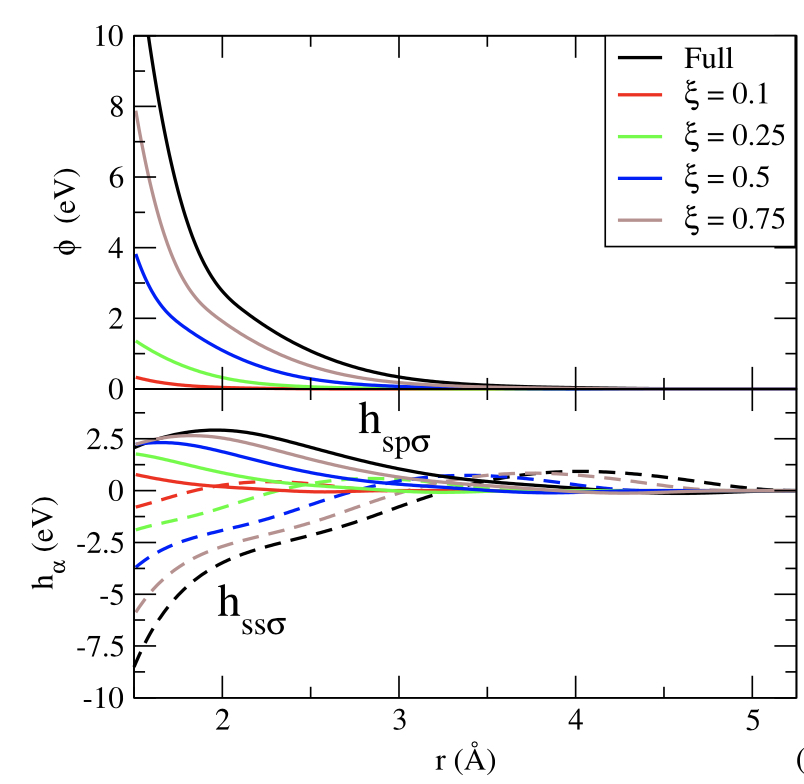
Evaluation of the grand-canonical partition function using expanded Wang-Landau simulations. IV. Performance of many-body force fields and tight-binding schemes for the fluid phases of silicon
C. Desgranges, J. Delhommelle
J. Chem. Phys., 144 p. 124510, 20162015
Many-Body Effects on the Thermodynamics of Fluids, Mixtures, and Nanoconfined Fluids
C. Desgranges, J. Delhommelle
J. Chem. Theory Comput., 11, p. 5401-5414, 2015
A new force field for H2S and its binary and ternary mixtures with CO2 and CH4
A.N. Owen, C. Desgranges, J. Delhommelle
Fluid Phase Equilibr., 402, p. 69-77, 20152014
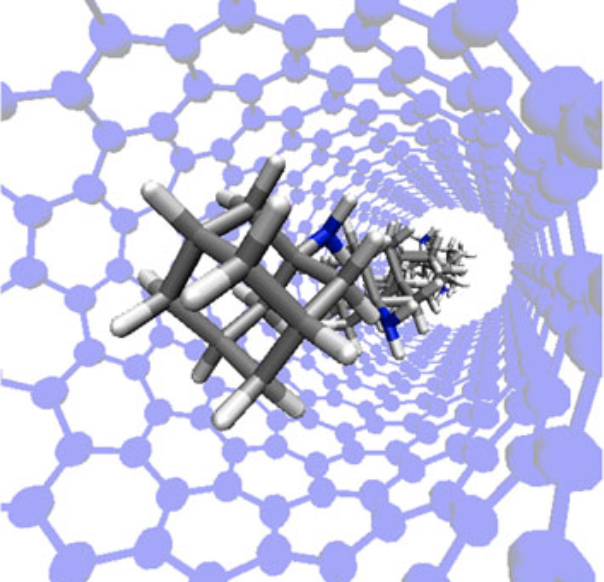
Adsorption and diffusion of the antiparkinsonian drug amantadine in carbon nanotubes
E. Hicks, C. Desgranges, J. Delhommelle
Molec. Simulat., 40, p. 656-663, 2014
Evaluation of the grand-canonical partition function using expanded Wang-Landau simulations. III. Impact of combining rules on mixtures properties
C. Desgranges, J. Delhommelle
J. Chem. Phys., 140, p. 104109, 2014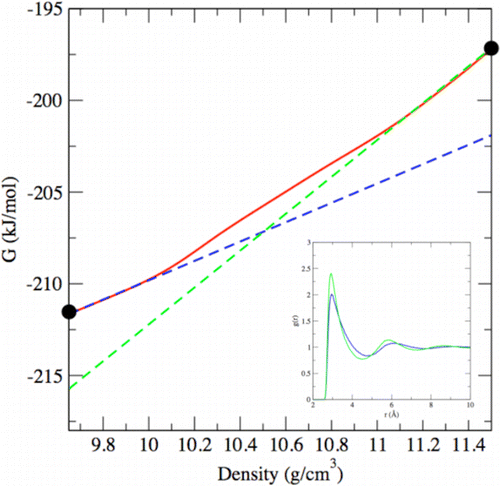
Thermodynamics of Phase Coexistence and Metal–Nonmetal Transition in Mercury: Assessment of Effective Potentials via Expanded Wang–Landau Simulations
C. Desgranges, J. Delhommelle
J. Phys. Chem. B, 118, p. 3175-3182, 2014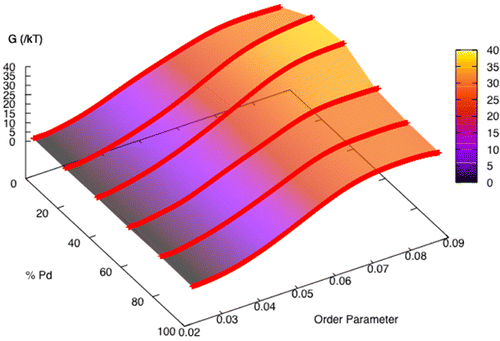
Unraveling the Coupling between Demixing and Crystallization in Mixtures
C. Desgranges, J. Delhommelle
J. Am. Chem. Soc., 136, p. 8145-8148, 2014
Adsorption of hydrogen in covalent organic frameworks using expanded Wang–Landau simulations
A.R.V. Koenig, C. Desgranges, J. Delhommelle
Molec. Simulat., 40, p. 71-79, 20142012
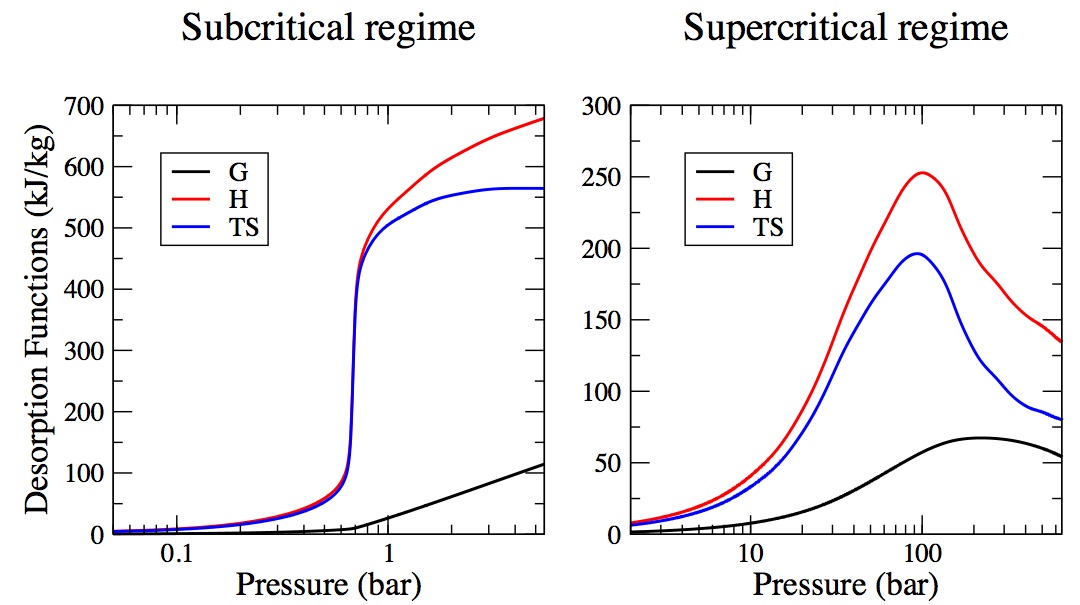
Characterization and Comparison of the Performance of IRMOF-1, IRMOF-8, and IRMOF-10 for CO2 Adsorption in the Subcritical and Supercritical Regimes
J. M. Hicks, C. Desgranges, J. Delhommelle
J. Phys. Chem. C, 116, p. 22938-22946, 2012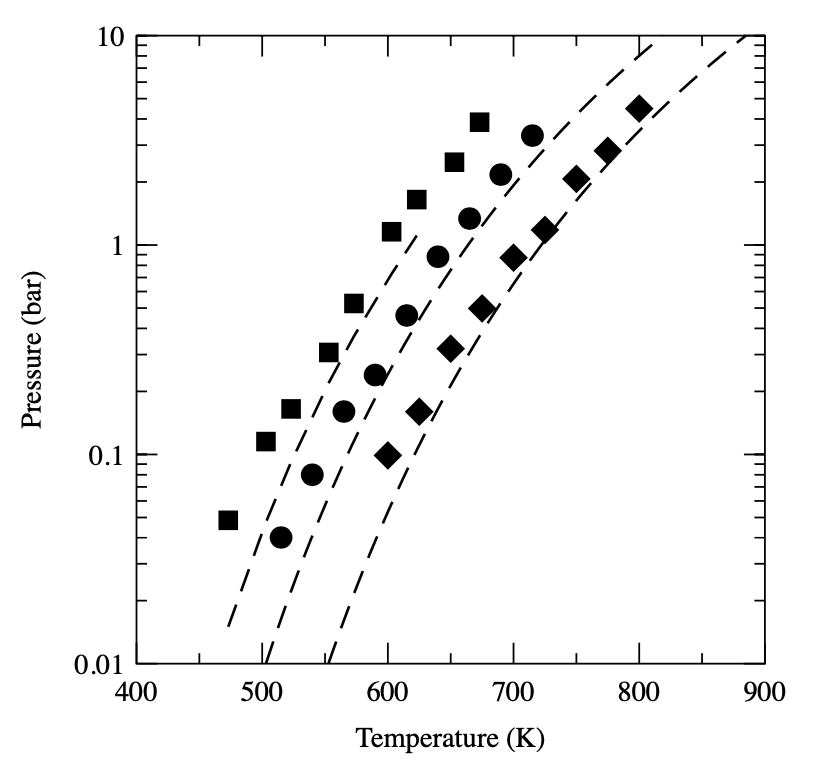
Numerical estimate for boiling points via Wang–Landau simulations
T. Aleksandrov, C. Desgranges, J. Delhommelle
Molec. Simulat., 38, p. 1265-1270, 2012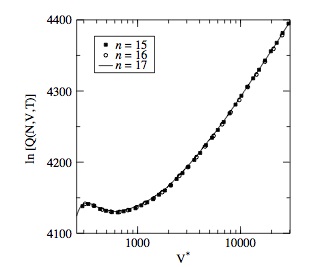
Wang–Landau configurational bias Monte Carlo simulations: vapour–liquid equilibria of alkenes
K. Ndumbe Ngale, C. Desgranges, J. Delhommelle
Molec. Simulat., 38, p. 653-658, 2012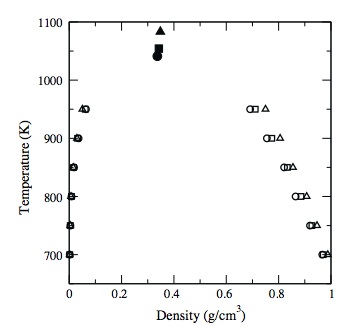
Prediction of critical properties for naphthacene, triphenylene and chrysene by Wang–Landau simulations
C. Desgranges, K. Ndumbe Ngale, J. Delhommelle
Fluid Phase Equilibr., 332, p. 92-96, 2012
Evaluation of the grand-canonical partition function using expanded Wang-Landau simulations. I. Thermodynamic properties in the bulk and at the liquid-vapor phase boundary
C. Desgranges, J. Delhommelle
J. Chem. Phys., 136, p. 184107, 2012
Evaluation of the grand-canonical partition function using expanded Wang-Landau simulations. II. Adsorption of atomic and molecular fluids in a porous material
C. Desgranges, J. Delhommelle
J. Chem. Phys., 136, p. 184108, 20122011
Polymorph selection during the crystallization of iron under the conditions of Earth’s inner core
J. Persson, C. Desgranges, J. Delhommelle
Chem. Phys. Lett., 511, p. 57-61, 2011
Crystal nucleation and growth from supercooled melts
J. Delhommelle
Molec. Simulat., 37, p. 613-620, 2011
Role of liquid polymorphism during the crystallization of Silicon
C. Desgranges, J. Delhommelle
J. Am. Chem. Soc., 133, p. 2872-2874, 2011
Crystal nucleation and growth in Pd–Ni alloys: a molecular simulation study
K. Watson, S.E. Tatsinkou Nguelo, C. Desgranges, J. Delhommelle
CrystEngComm, 13, p. 1132-1140, 20112010
Optimisation of multiple time-step hybrid Monte Carlo Wang–Landau simulations in the isobaric–isothermal ensemble for the determination of phase equilibria
C. Desgranges, E.A. Kastl, T. Aleksandrov, J. Delhommelle
Molec. Simulat., 36, p. 544-551, 2010
Phase equilibria of polyaromatic hydrocarbons by hybrid Monte Carlo Wang–Landau simulations
C. Desgranges, J.M. Hicks, A. Magness, J. Delhommelle
Mol. Phys., 108, p. 151-158, 2010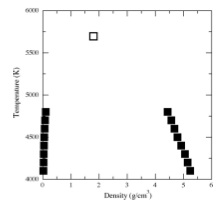
Vapor–liquid equilibria of copper using hybrid Monte Carlo Wang—Landau simulations
T. Aleksandrov, C. Desgranges, J. Delhommelle
Fluid Phase Equilibr., 287, p. 79-83, 20102009
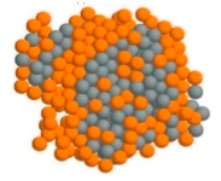
Nucleation and growth of C60 nanoparticles from the supersaturated vapor and from the undercooled liquid: A molecular simulation study
K. Ndumbe Ngale, C. Desgranges, J. Delhommelle
J. Chem. Phys., 131, p. 244515, 2009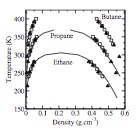
Phase equilibria of molecular fluids via hybrid Monte Carlo Wang–Landau simulations: Applications to benzene and n-alkanes
C. Desgranges, J. Delhommelle
J. Chem. Phys., 130, p. 244109, 2009
Universal scaling law for energy and pressure in a shearing fluid
C. Desgranges, J. Delhommelle
Phys. Rev. E, 79, p. 052201, 2009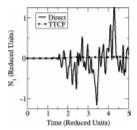
Accurate determination of normal stress differences via transient-time correlation function–non-equilibrium molecular dynamics (TTCF–NEMD) simulations
C. Desgranges, J. Delhommelle
Molec. Simulat., 35, p. 405-408, 2009
Molecular simulation of the nucleation and growth of gold nanoparticles
C. Desgranges, J. Delhommelle
J. Phys. Chem. C, 113, p. 3607-3611, 20092008
Rheology of liquid fcc metals: Equilibrium and transient-time correlation-function nonequilibrium molecular dynamics simulations
C. Desgranges, J. Delhommelle
Phys. Rev. B, 78, p. 184202, 2008
Shear viscosity of liquid copper at experimentally accessible shear rates: Application of the transient-time correlation function formalism
C. Desgranges, J. Delhommelle
J. Chem. Phys., 128, p. 084506, 2009
Molecular simulation of transport in nanopores: Application of the transient-time correlation function formalism
C. Desgranges, J. Delhommelle
Phys. Rev. E, 77, p. 027701, 2008
Crystallization mechanisms for supercooled liquid Xe at high pressure and temperature: Hybrid Monte Carlo molecular simulations
C. Desgranges, J. Delhommelle
Phys. Rev. B, 77, p. 054201, 2008
Estimating the conductivity of a nanoconfined liquid subjected to an experimentally accessible external field
C. Desgranges, J. Delhommelle
Molec. Simulat., 34, p. 177-181, 20092007
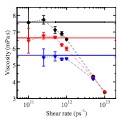
Viscosity of liquid iron under high pressure and high temperature: Equilibrium and nonequilibrium molecular dynamics simulation studies
C. Desgranges, J. Delhommelle
Phys. Rev. B, 76, p. 172102, 2007
Polymorph selection during the crystallization of softly repulsive spheres: The inverse power law potential
C. Desgranges, J. Delhommelle
J. Phys. Chem. B, 111, p. 12257-12262, 2007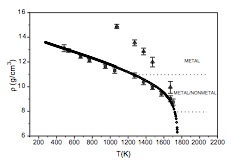
Structure and thermodynamics of the expanded liquid mercury by Monte Carlo simulation: a first attempt
J.-M. Bomont, J. Delhommelle, J.-L. Bretonnet
J. Non-Cryst. Solids, 353, p. 3454-3458, 2007
Molecular simulation of the crystallization of aluminum from the supercooled liquid
C. Desgranges, J. Delhommelle
J. Chem. Phys., 127, p. 144509, 2007
Hit and miss of classical nucleation theory as revealed by a molecular simulation study of crystal nucleation in supercooled sulfur hexafluoride
J.-M. Leyssale, J. Delhommelle, C. Millot
J. Chem. Phys., 127, p. 044504, 2007
Controlling polymorphism during the crystallization of an atomic fluid
C. Desgranges, J. Delhommelle
Phys. Rev. Lett., 98, p. 235502, 2007
Molecular insight into the pathway to crystallization of aluminum
C. Desgranges, J. Delhommelle
J. Am. Chem. Soc., 129, p. 7012-7013, 2007
Molecular simulation of cross-nucleation between polymorphs
C. Desgranges, J. Delhommelle
J. Phys. Chem. B, 111, p. 1465-1469, 2007
Polymorph selection during the crystallization of Yukawa systems
C. Desgranges, J. Delhommelle
J. Chem. Phys., 126, p. 054501, 20072006
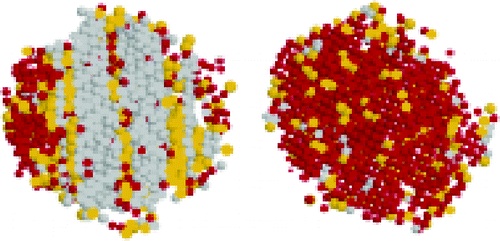
Insights into the molecular mechanism underlying polymorph selection
C. Desgranges, J. Delhommelle
J. Am. Chem. Soc., 128, p. 15104-15105, 2006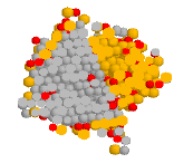
Molecular mechanism for the cross-nucleation between polymorphs
C. Desgranges, J. Delhommelle
J. Am. Chem. Soc., 128, p. 10368-10369, 20062005
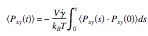
Simulation of friction in nanoconfined fluids for an arbitrarily low shear rate
J. Delhommelle, P.T. Cummings
Phys. Rev. B., 72, p. 172201, 2005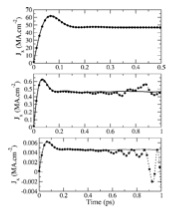
Conductivity of molten sodium chloride in an arbitrarily weak dc electric field
J. Delhommelle, P.T. Cummings, J. Petravic
J. Chem. Phys., 123, p. 114505, 2005
Shear thickening in a model colloidal suspension
J. Delhommelle, J. Petravic
J. Chem. Phys., 123, p. 074707, 2005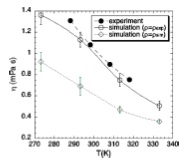
Hydrogen bonding in ethanol under shear
J. Petravic, J. Delhommelle
J. Chem. Phys., 122, p. 234509, 2005
Molecular simulation of the homogeneous crystal nucleation of carbon dioxide
J.-M. Leyssale, J. Delhommelle, C. Millot
J. Chem. Phys., 122, p. 184518, 2005
Atomistic simulation of the homogeneous nucleation and of the growth of N2 crystallites
J.-M. Leyssale, J. Delhommelle, C. Millot
J. Chem. Phys., 122, p. 104510, 2005
Should “lane formation” occur systematically in driven liquids and colloids?
J. Delhommelle
Phys. Rev. E, 71, p. 016705, 20052004
Reorganization and growth of metastable α-N2 critical nuclei into stable β-N2 crystals
J.-M. Leyssale, J. Delhommelle, C. Millot
J. Am. Chem. Soc., 126, p. 12286-12287, 2004
Nonequilibrium molecular dynamics simulations of molten sodium chloride
J. Petravic, J. Delhommelle
Int. J. Thermophysics, 25, p. 1375-1393, 2004
Simulations of shear-induced melting in two dimensions
J. Delhommelle
Phys. Rev. B, 69, p. 144117, 2004
Non-Newtonian behavior in simple fluids
J. Delhommelle, J. Petravic, D.J. Evans
J. Chem. Phys., 120, p. 6117-6123, 20042003
On the effects of assuming flow profiles in nonequilibrium simulations
J. Delhommelle, J. Petravic, D.J. Evans
J. Chem. Phys., 119, p. 11005-11010, 2003
Conductivity of molten sodium chloride in an alternating electric field
J. Petravic, J. Delhommelle
J. Chem. Phys., 119, p. 8511-8518, 2003
Shear viscosity of molten sodium chloride
J. Delhommelle, J. Petravic
J. Chem. Phys., 118, p. 2783-2791, 2003
Influence of temperature, pressure and internal degrees of freedom on hydrogen bonding and diffusion in liquid ethanol
J. Petravic, J. Delhommelle
Chem. Phys., 286, p. 303-314, 20032002
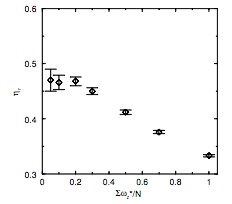
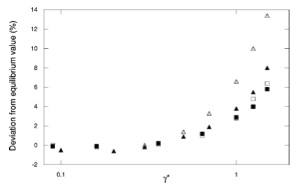
Correspondence between configurational temperature and molecular kinetic temperature thermostats
J. Delhommelle, D.J. Evans
J. Chem. Phys., 117, p. 6016-6021, 2002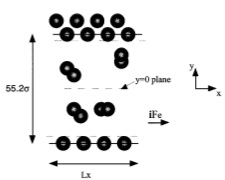
Poiseuille flow of a micropolar fluid
J. Delhommelle, D.J. Evans
Mol. Phys., 100, p. 2857-2865, 2002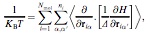
Configurational thermostats for molecular systems
L. Lue, O.G. Jepps, J. Delhommelle, D.J. Evans
Mol. Phys., 100, p. 2387-2395, 20022001
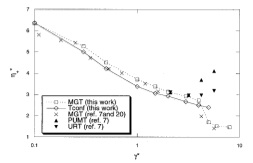
Configurational temperature thermostat for fluids undergoing shear flow: application to liquid chlorine
J. Delhommelle, D.J. Evans
Mol. Phys., 99, p. 1825-1829, 2001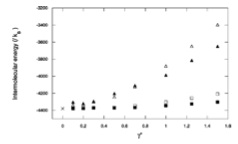
Comparison of thermostatting mechanisms in NVT and NPT simulations of decane under shear
J. Delhommelle, D.J. Evans
J. Chem. Phys., 115, p. 43-49, 2001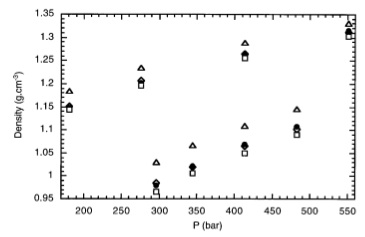
Inadequacy of the Lorentz-Berthelot combining rules for accurate predictions of equilibrium properties by molecular simulation
J. Delhommelle, P. Millie
Mol. Phys., 99, p. 619-625, 2001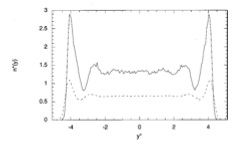
Configurational temperature profile in confined fluids. II. Molecular fluids
J. Delhommelle, D.J. Evans
J. Chem. Phys., 104, p. 6236-6241, 2001
Configurational temperature profile in confined fluids. I. Atomic fluid
J. Delhommelle, D.J. Evans
J. Chem. Phys., 114, p. 6229-6235, 20012001
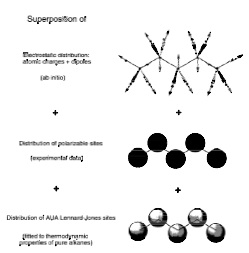
On the role of the definition of potential models in Gibbs ensemble phase equilibria simulations of the H2S-pentane mixture
J. Delhommelle, P. Millie, A.H. Fuchs
Mol. Phys., 98, p. 1895-1905, 2000
Derivation of an optimized potential model for phase equilibria (OPPE) for sulfides and thiols
J. Delhommelle, C. Tschirwitz, Ph. Ungerer, G. Granucci, P. Millie, D. Pattou, A.H. Fuchs
J. Phys. Chem. B, 104, p. 4745-4753, 2000
Optimization of the anisotropic united atoms intermolecular potential for n-alkanes
Ph. Ungerer, C. Beauvais, J. Delhommelle, A. Boutin, B. Rousseau, A.H. Fuchs
J. Chem. Phys., 112, p. 5499-5510, 2000
Etablissement de potentiels d'interaction pour la simulation moleculaire. Application a la prediction des equilibres liquide-vapeur de melanges binaires alcane-molecule multipolaire
J. Delhommelle
PhD Thesis, 20001999
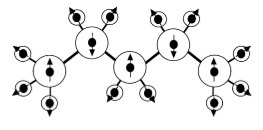
A new method for deriving atomic charges and dipoles for n,-alkanes: investigation of transferability and geometry dependence
J. Delhommelle, G. Granucci, V. Brenner, P. Millie, A. Boutin, A.H. Fuchs
Mol. Phys., 97, p. 1117-1128, 1999
Molecular simulation of vapour-liquid coexistence curves for hydrogen sulfide-alkane and carbon dioxide-alkane mixtures
J. Delhommelle, A. Boutin, A.H. Fuchs
Molec. Simulat., 22, p. 351-368, 1999
Vapour-liquid coexistence curves of the united-atom and anisotropic united-atom force fields for alkane mixtures
J. Delhommelle, A. Boutin, B. Travitian, A.D. Mackie, A.H. Fuchs
Mol. Phys., 96, p. 1517-1524, 1999
Monte Carlo simulations of squalane in the Gibbs ensemble
B. Neubauer, J. Delhommelle, A. Boutin, B. Travitian, A.H. Fuchs
Fluid Phase. Equilibr., 155, p. 167-176, 1999